FENYLKETONURIE (PKU)
První případy fenylketonurie byly zaznamenány již v roce 1934 u dvou sourozenců s těžkou mentální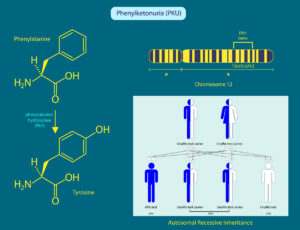 retardací. Důvod jejich postižení, poruchu metabolismu fenylalaninu, popsal norský lékař a biochemik Ivar Asbjørn Følling na základě izolace fenylpyruvátu z jejich moče. Pravou příčinu této choroby, mutaci jaterního enzymu fenylalaninhydroxylázy, určil mezi lety 1947 a 1953 lékař George Jervis. Od té doby bylo objeveno více než 280 mutací tohoto i dalších enzymů hrající roli ve vzniku onemocnění zvaném fenylketonurie.
retardací. Důvod jejich postižení, poruchu metabolismu fenylalaninu, popsal norský lékař a biochemik Ivar Asbjørn Følling na základě izolace fenylpyruvátu z jejich moče. Pravou příčinu této choroby, mutaci jaterního enzymu fenylalaninhydroxylázy, určil mezi lety 1947 a 1953 lékař George Jervis. Od té doby bylo objeveno více než 280 mutací tohoto i dalších enzymů hrající roli ve vzniku onemocnění zvaném fenylketonurie.
Fenylketonurie je dědičné metabolické onemocnění spočívající v poruše přeměny aminokyseliny fenylalaninu na tyrosin, jenž u zdravých lidí katalyzuje jaterní enzym fenylalaninhydroxyláza (PAH). Právě mutaci genu kódujícího tento enzym má největší procento pacientů s fenylketonurií. Fenylalanin se pak hromadí v krvi a tkáních a jakmile vznikne v organismu dostatečná „zásoba“ stává se pro člověka toxickou – poškozuje vývoj mozku a rozvíjí se mentální retardace. Fenylketonurie se diagnostikuje pomocí testu. Tento test spočívá v odběru kapky krve z patičky novorozence na testovací kartičku, která se odesílá do speciální laboratoře – novorozenecký screening. V ČR probíhá testování všech novorozenců ze zákona od roku 1975. Výskyt onemocnění se v ČR dle jednotlivých zdrojů různí, většinou je uváděna incidence 1:9 000, což odpovídá 12 nově diagnostikovaným případům ročně.
 Fenylketonurii zatím není možné vyléčit a je proto nutné dodržovat nízkobílkovinnou dietu. Omezení příjmu bílkovin spolu s medicínskou podporou, která spočívá především v dodávání vhodných aminokyselinových směsí bez fenylalaninu a dodávání dalších látek a vitaminů, je nejčastější a většinou také jedinou možností jak zamezit poškození organizmu. Pacienti s PKU však dnes již mohou žít plnohodnotným životem lišícím se od životů svých vrstevníků pouze rozdílnou stravou.
Fenylketonurii zatím není možné vyléčit a je proto nutné dodržovat nízkobílkovinnou dietu. Omezení příjmu bílkovin spolu s medicínskou podporou, která spočívá především v dodávání vhodných aminokyselinových směsí bez fenylalaninu a dodávání dalších látek a vitaminů, je nejčastější a většinou také jedinou možností jak zamezit poškození organizmu. Pacienti s PKU však dnes již mohou žít plnohodnotným životem lišícím se od životů svých vrstevníků pouze rozdílnou stravou.